 Vulcan Pipeline
Vulcan PipelineBackground
Long-read sequencing has enabled unprecedented surveys of structural variation across the entire human genome. To maximize the potential of long-read sequencing in this context, novel mapping methods have emerged that have primarily focused on either speed or accuracy. Various heuristics and scoring schemas have been implemented in widely used read mappers (minimap2 and NGMLR) to optimize for speed or accuracy, which have variable performance across different genomic regions and for specific structural variants. Our hypothesis is that constraining read mapping to the use of a single gap penalty across distinct mutational hot spots reduces read alignment accuracy and impedes structural variant detection.
Findings
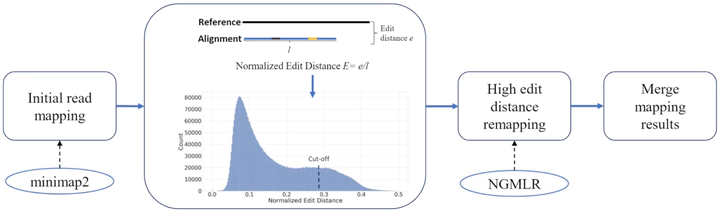
We tested our hypothesis by implementing a read-mapping pipeline called Vulcan that uses two distinct gap penalty modes, which we refer to as dual-mode alignment. The high-level idea is that Vulcan leverages the computed normalized edit distance of the mapped reads via minimap2 to identify poorly aligned reads and realigns them using the more accurate yet computationally more expensive long-read mapper (NGMLR). In support of our hypothesis, we show that Vulcan improves the alignments for Oxford Nanopore Technology long reads for both simulated and real datasets. These improvements, in turn, lead to improved accuracy for structural variant calling performance on human genome datasets compared to either of the read-mapping methods alone.
Conclusions
Vulcan is the first long-read mapping framework that combines two distinct gap penalty modes for improved structural variant recall and precision. Vulcan is open-source and available under the MIT License at https://gitlab.com/treangenlab/vulcan.
Collaborators
- Dr. Fritz Sedlazeck (BCM HGSC)
- Dr. Medhat Mahmoud (BCM HGSC)
Dr. Yilei Fu
PhD student from September 2019 through January 2024
Dr. Fu is currently a postdoctoral scientist in the Sedlazeck lab at BCM HGSC. Dr. Fu received his Ph.D. from Rice University in December 2023. Previously, Yilei (4th year PhD student) obtained B.E. degrees in Computer Science and Technology from Harbin Institute of Technology, China. During his undergraduate procedure, he receive National People’s Scholorship for 3 times. In the meantime, he also worked on detecting SV(structure variation) on human genomes using third generation sequencing data. His research interest can be briefly discribed as exploring gene-carried information using third/next generation sequencing data.
Todd J. Treangen
Associate Professor of Computer Science, Bioengineering
My research interests include algorithms and data structures for efficient analysis of microbial genomes and metagenomes